

《易經》第二十卦‧風地觀
觀:盥而不荐,有孚顒若。
彖曰:大觀在上,順而巽,中正以觀天下。觀,盥而不荐,有孚顒若,下觀而化也。 觀天之神道 ,而四時不忒, 聖人以神道設教 ,而天下服矣。
象曰:風行地上,觀﹔先王以省方,觀民設教。
初六:童觀,小人無咎,君子吝 。
象曰:初六童觀,小人道也。
六二:窺觀,利女貞。
象曰:窺觀女貞,亦可丑也。
六三:觀我生,進退。
象曰:觀我生,進退﹔未失道也。
六四:觀國之光,利用賓于王。
象曰:觀國之光,尚賓也。
九五:觀我生,君子無咎。
象曰:觀我生,觀民也。
上九:觀其生,君子無咎。
象曰:觀其生,志未平也。
《論語‧學而篇》
子曰:不患人之不己知,患不知人也。
《論語 ‧為政篇》
子曰:視其所以,觀其所由,察其所安;人焉廋哉。人焉廋哉。
…
【若果】『一葉知秋』,履霜繼至堅冰,所以或有『深謀遠慮』,善採『先天下之憂而憂』之『舉措之旨』。所謂的『大觀在上』是說『自然人生法則』,『順而巽』是講『順應自然律』而且『深入其理』,這就是『觀』之大者!!
……
天地之《生》是一,《生生》故為多。所謂『數』始於一,然後二,然後三,然後很多很多。恰似古今中外,都有個『函三』『惟‧唯』一的說法︰
西︰ 聖父,聖子,聖靈
東︰ 太易、太初、太始
於是千百億年之後,『眾生』蔚為『大觀』,有人開始用『理性』之『眼』,欲觀宇宙『大霹靂』之『開天闢地』的『溫柔』。尋找元初的
立德,立功,立言
,卻是
不見其『德』,有『道』
難覓其『功』,成『果』
不知其『言』,現『象』
,當真『不識廬山真面目』的耶!
因此現今有人欲察『派生』之『博多』,當如何求『其一』的呢?『物』有『始』『中』『終』,或求『其始』的吧︰
『東西』 Object 為本!
─── 《W!O 的派生‧十日談之《八》》
『養身』可以『養生』耶?故耳黃帝問『風』也!
《黃帝內經‧素問》風論篇第四十二
黃帝問曰:風之傷人也,或為寒熱,或為熱中,或為寒中,或為癘風,或為偏枯,或為風也,其病各異,其名不同。或內至五臟六腑 ,不知其解,願聞其說。
岐伯對曰:風氣藏在皮膚之間,內不得通,外不得洩。風者,善行而數變,腠理開,則灑然寒,閉則熱而悶。其寒也,則衰食飲;其熱也,則消肌肉。故使人怢慄而不能食,名曰寒熱。風氣與陽明入胃,循脈而上至目內眥,其人肥,則風氣不得外洩,則為熱中而目黃;人瘦則外洩而寒,則為寒中而泣出。風氣與太陽俱入,行諸脈俞,散於分肉之間,與衛氣相干,其道不利。故使肌肉憤●而有瘍,衛氣有所凝而不行,故其肉有不仁也。癘者,有榮氣熱腑,其氣不清,故使其鼻柱壞而色敗,皮膚瘍潰。風寒客於脈而不去,名曰癘風,或名曰寒熱。以春甲乙傷於風者為肝風,以夏丙丁傷於風者為心風,以季夏戊己傷於邪者為脾風,以秋庚辛中於邪者為肺風,以冬壬癸中於邪者為腎風。風中五臟六腑之俞,亦為臟腑之風,各入其門戶,所中則為偏風。風氣循風府而上,則為胸風,風入系頭,則為目風,眼寒。飲酒中風,則為漏風。入房汗出中風,則為內風 。新沐中風,則為首風。久風入中,則為腸風,飧洩。外在腠理,則為洩風。故風者,百病之長也,至其變化,乃為他病也,無常方 ,然致有風氣也。
………
所謂
風邪
風邪是中醫學上對一類外界環境致病因素的稱呼,為六淫之一。《黃帝內經》中描述為:「風者,善行而數變。」[1]。風邪為陽邪 ,侵襲部位多在表、在上,具有遊走性,病情變化較多且較迅速 。感受風邪的常見症狀有頭痛、寒熱汗出、遍身遊走疼痛和肌膚瘙癢等。風邪常與其他病邪結合一同致病,如風寒、風熱、風濕、風燥等。
者,內外門戶乎??
因是知『平衡』
Chemical equilibrium
In a chemical reaction, chemical equilibrium is the state in which both reactants and products are present in concentrations which have no further tendency to change with time, so that there is no observable change in the properties of the system.[1] Usually, this state results when the forward reaction proceeds at the same rate as the reverse reaction. The reaction rates of the forward and backward reactions are generally not zero, but equal. Thus, there are no net changes in the concentrations of the reactant(s) and product(s). Such a state is known as dynamic equilibrium.[2][3]
仰賴『環境』呦!!
Historical introduction
The concept of chemical equilibrium was developed after Berthollet (1803) found that some chemical reactions are reversible.[4] For any reaction mixture to exist at equilibrium, the rates of the forward and backward (reverse) reactions are equal. In the following chemical equation with arrows pointing both ways to indicate equilibrium,[5] A and B are reactant chemical species, S and T are product species, and α, β, σ, and τ are the stoichiometric coefficients of the respective reactants and products:
- α A + β B ⇌ σ S + τ T
The equilibrium concentration position of a reaction is said to lie “far to the right” if, at equilibrium, nearly all the reactants are consumed. Conversely the equilibrium position is said to be “far to the left” if hardly any product is formed from the reactants.
Guldberg and Waage (1865), building on Berthollet’s ideas, proposed the law of mass action:
質量作用定律
質量作用定律(英語:Law of mass action)是化學領域的概念。定律包含兩個方向:
- 平衡方向,關於平衡時的反應混合物的組成。
- 動力學方向,關於基元反應的速率式。
表述
對於一個簡單地基元反應:  ,記化學反應速率為
,記化學反應速率為  ,則有:
,則有: ![\displaystyle v=k[X]^{a}[Y]^{b}](https://www.freesandal.org/wp-content/ql-cache/quicklatex.com-cb72563e577ab88a888a2f3fe27647ec_l3.png "Rendered by QuickLaTeX.com") 。其中[X]表示反應物X的濃度,k表示反應的速率常數。
。其中[X]表示反應物X的濃度,k表示反應的速率常數。
作為對動力學的表述:一個基元反應[1]的速率,是參與反應的分子的濃度的乘積的比例。 在近代化學中,這是用統計力學推導出來的。
而作為對平衡的陳述,這個定律給出了平衡常數的關係式,平衡常數是可以描述化學平衡的一個量。在近代化學中,這是由平衡熱力學推導出來的。

where A, B, S and T are active masses and k+ and k− are rate constants. Since at equilibrium forward and backward rates are equal:

and the ratio of the rate constants is also a constant, now known as an equilibrium constant.

By convention the products form the numerator. However, the law of mass action is valid only for concerted one-step reactions that proceed through a single transition state and is not valid in general because rate equations do not, in general, follow the stoichiometry of the reaction as Guldberg and Waage had proposed (see, for example, nucleophilic aliphatic substitution by SN1 or reaction of hydrogen and bromine to form hydrogen bromide). Equality of forward and backward reaction rates, however, is a necessary condition for chemical equilibrium, though it is not sufficient to explain why equilibrium occurs.
Despite the failure of this derivation, the equilibrium constant for a reaction is indeed a constant, independent of the activities of the various species involved, though it does depend on temperature as observed by the van ‘t Hoff equation. Adding a catalyst will affect both the forward reaction and the reverse reaction in the same way and will not have an effect on the equilibrium constant. The catalyst will speed up both reactions thereby increasing the speed at which equilibrium is reached.[2][6]
Although the macroscopic equilibrium concentrations are constant in time, reactions do occur at the molecular level. For example, in the case of acetic acid dissolved in water and forming acetate and hydronium ions,
- CH3CO2H + H2O ⇌ CH3CO−2 + H3O+
a proton may hop from one molecule of acetic acid on to a water molecule and then on to an acetate anion to form another molecule of acetic acid and leaving the number of acetic acid molecules unchanged. This is an example of dynamic equilibrium. Equilibria, like the rest of thermodynamics, are statistical phenomena, averages of microscopic behavior.
Le Châtelier’s principle (1884) gives an idea of the behavior of an equilibrium system when changes to its reaction conditions occur. If a dynamic equilibrium is disturbed by changing the conditions, the position of equilibrium moves to partially reverse the change. For example, adding more S from the outside will cause an excess of products, and the system will try to counteract this by increasing the reverse reaction and pushing the equilibrium point backward (though the equilibrium constant will stay the same).
If mineral acid is added to the acetic acid mixture, increasing the concentration of hydronium ion, the amount of dissociation must decrease as the reaction is driven to the left in accordance with this principle. This can also be deduced from the equilibrium constant expression for the reaction:

If {H3O+} increases {CH3CO2H} must increase and CH3CO−2 must decrease. The H2O is left out, as it is the solvent and its concentration remains high and nearly constant.
A quantitative version is given by the reaction quotient.
J. W. Gibbs suggested in 1873 that equilibrium is attained when the Gibbs free energy of the system is at its minimum value (assuming the reaction is carried out at constant temperature and pressure). What this means is that the derivative of the Gibbs energy with respect to reaction coordinate (a measure of the extent of reaction that has occurred, ranging from zero for all reactants to a maximum for all products) vanishes, signalling a stationary point.
反應坐標
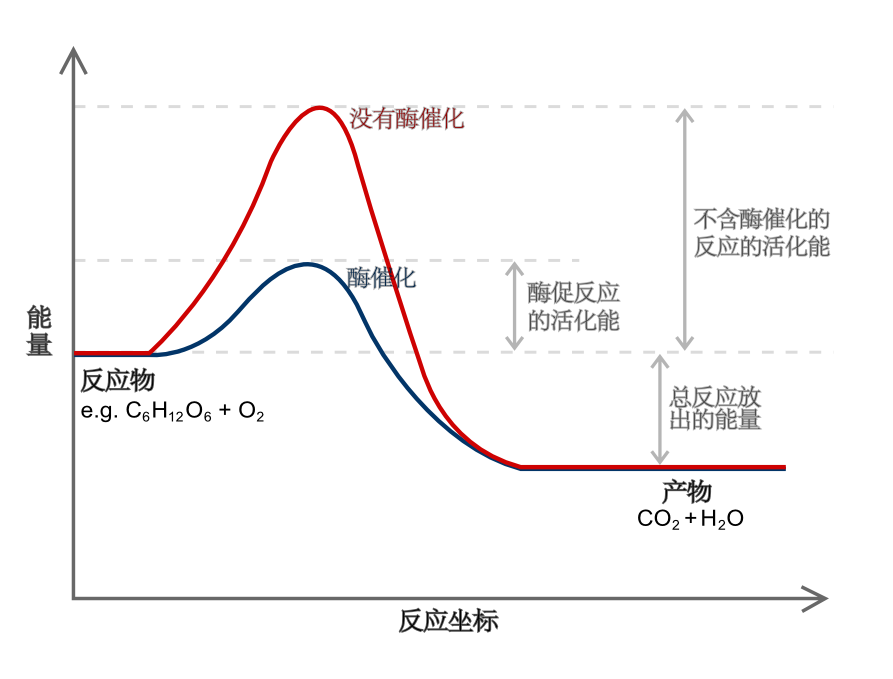
圖示為一個反應坐標,當中位於能量最高點的均為過渡態。紅色線表示了沒有加入催化劑(酶)因此具有較高的活化能,藍色線是加了催化劑後的情況。
反應坐標,或稱作能線圖是一種根據反應途徑加以圖像化的一維坐標,用以表達化學反應的進行過程[1]。利用簡單的幾何圖表,加以表示單個或數個分子實體在化學反應中所經歷轉變的途徑。
這類坐標有時也可用鍵長、鍵角等參數來表示,但最常見的則是用自由能作為主要參數。一些非幾何參數有時會在更複雜的反應中使用,例如是不同分子實體的鍵級。
在過渡態理論中,反應坐標就是一組以曲線組成的坐標,用以表示某種化學實體由本身的結構,經變化成過渡態,最後演變成某結構的生成物的整個過程。在坐標中上升斜率最大的一個途徑,就是該反應的速率控制步驟。
反應進度
反應進度(英文:Extent of reaction)是用於量化反映化學反應進行程度的化學量,在運算中通常以希臘字母ξ(Xi)代表。一般反應進度的單位為摩爾。一個進行中或已完成的化學反應,無論計算哪一個反應物或生成物的反應程度,都會得到相同的結果,因為反應進度代表整個反應的進度,而不是單一物質的反應進度。
基本定義
考慮一化學反應計量式:
- A ⇌ B
這個化學反應計量式表示化學物質A在特定條件下等量地與化學物質B相互轉換。假設有無限少量的A物質透過此反應生成B物質,則 A 物質數量的變化量為 dnA=-dξ ,而 B 物質數量的變化量為 dnB=dξ[1] ,則反應程度 ξ 將定義為:[2][3] 更一般地,若化學反應計量式中,第i個化學物質(反應物或生成物)的化學計量係數是  ,其物質的量為
,其物質的量為  ,則
,則

其中,需要特別注意的是 值的正負號。選定了反應的正方向後,當選取的物質為反應物時, 為負值;若為生成物,則為正值。唯有如此規範,才能使反應程度 ξ 之值恆正。
若非無限少量的反應,而是有限變化量的反應,則可將上式通過對時間積分而改寫為:

若假設 t=0 時,反應進度 ξ=0,則反應進度ξ可表示為:

This derivative is called the reaction Gibbs energy (or energy change) and corresponds to the difference between the chemical potentials of reactants and products at the composition of the reaction mixture.[1] This criterion is both necessary and sufficient. If a mixture is not at equilibrium, the liberation of the excess Gibbs energy (or Helmholtz energy at constant volume reactions) is the “driving force” for the composition of the mixture to change until equilibrium is reached. The equilibrium constant can be related to the standard Gibbs free energy change for the reaction by the equation

where R is the universal gas constant and T the temperature.
When the reactants are dissolved in a medium of high ionic strength the quotient of activity coefficients
活度係數
活度係數(英語:Activity coefficient),又稱活度因子(英語 :Activity factor),是熱力學中的一個係數,反映的是真實溶液中某組分i的行為偏離理想溶液的程度[1],量綱為1。引入活度係數後 ,適用於理想溶液的各種關係可以相應修正為適用於真實溶液。類似的,逸度係數是表示真實氣體混合物中某組分和理想行為的偏離的係數。
定義
在理想溶液中,溶液組分 i 遵循拉午耳定律:

其中  是組分 i 在溶液中的莫耳分率,
是組分 i 在溶液中的莫耳分率,  和
和  分別是組分 i 的分壓和飽和蒸氣壓。 而組分 i 的化學勢
分別是組分 i 的分壓和飽和蒸氣壓。 而組分 i 的化學勢  可由下式表達:
可由下式表達:

這裡的  代表組分 i 在標準狀態下的化學勢。而在真實溶液中,組分 i-組分 i 間的作用力和組分 i-其他組分間的作用力並不相等,導致了組分i並不滿足拉午耳定律,其化學勢也不滿足以上關係,即偏離了理想溶液的行為,為此吉爾伯特·牛頓·路易士引入了活度和活度係數的概念。 定義:
代表組分 i 在標準狀態下的化學勢。而在真實溶液中,組分 i-組分 i 間的作用力和組分 i-其他組分間的作用力並不相等,導致了組分i並不滿足拉午耳定律,其化學勢也不滿足以上關係,即偏離了理想溶液的行為,為此吉爾伯特·牛頓·路易士引入了活度和活度係數的概念。 定義:

這裡的  是組分 i 以莫耳分率所表示的活度,
是組分 i 以莫耳分率所表示的活度, 則是組分i用莫耳分率所表示的活度係數。引入活度和活度係數後,拉午耳定律可以修正為:
則是組分i用莫耳分率所表示的活度係數。引入活度和活度係數後,拉午耳定律可以修正為:

組分i的化學勢則可以修正為:

真實溶液的濃度越稀,溶劑的活度係數就越接近1,活度和莫耳分率近乎相等,其行為越接近理想溶液。濃度越高,活度係數越偏離1,真實溶液的行為偏差理想溶液就越大,比如對於濃度較高的電解質溶液,其活度就無法用莫耳分率取代,這一點在電化學和土壤化學中十分常見[2]。
may be taken to be constant. In that case the concentration quotient, Kc,
![\displaystyle K_{\ce {c}}={\frac {[{\ce {S}}]^{\sigma }[{\ce {T}}]^{\tau }}{[{\ce {A}}]^{\alpha }[{\ce {B}}]^{\beta }}}](https://www.freesandal.org/wp-content/ql-cache/quicklatex.com-c08955098eb310b05a748b2757f70dd6_l3.png "Rendered by QuickLaTeX.com")
where [A] is the concentration of A, etc., is independent of the analytical concentration of the reactants. For this reason, equilibrium constants for solutions are usually determined in media of high ionic strength. Kc varies with ionic strength, temperature and pressure (or volume). Likewise Kp for gases depends on partial pressure. These constants are easier to measure and encountered in high-school chemistry courses.
Thermodynamics
At constant temperature and pressure, one must consider the Gibbs free energy, G, while at constant temperature and volume, one must consider the Helmholtz free energy: A, for the reaction; and at constant internal energy and volume, one must consider the entropy for the reaction: S.
The constant volume case is important in geochemistry and atmospheric chemistry where pressure variations are significant. Note that, if reactants and products were in standard state (completely pure), then there would be no reversibility and no equilibrium. Indeed, they would necessarily occupy disjoint volumes of space. The mixing of the products and reactants contributes a large entropy (known as entropy of mixing) to states containing equal mixture of products and reactants. The standard Gibbs energy change, together with the Gibbs energy of mixing, determine the equilibrium state.[7][8]
In this article only the constant pressure case is considered. The relation between the Gibbs free energy and the equilibrium constant can be found by considering chemical potentials.[1]
At constant temperature and pressure, the Gibbs free energy, G, for the reaction depends only on the extent of reaction: ξ (Greek letter xi), and can only decrease according to the second law of thermodynamics. It means that the derivative of G with ξ must be negative if the reaction happens; at the equilibrium the derivative being equal to zero.
 : equilibrium
: equilibrium
In order to meet the thermodynamic condition for equilibrium, the Gibbs energy must be stationary, meaning that the derivative of G with respect to the extent of reaction: ξ, must be zero. It can be shown that in this case, the sum of chemical potentials of the products is equal to the sum of those corresponding to the reactants. Therefore, the sum of the Gibbs energies of the reactants must be the equal to the sum of the Gibbs energies of the products.

where μ is in this case a partial molar Gibbs energy, a chemical potential. The chemical potential of a reagent A is a function of the activity, {A} of that reagent.

(where μoA is the standard chemical potential).
The definition of the Gibbs energy equation interacts with the fundamental thermodynamic relation to produce
 .
.
Inserting dNi = νi dξ into the above equation gives a Stoichiometric coefficient ( ) and a differential that denotes the reaction occurring once (dξ). At constant pressure and temperature the above equations can be written as
) and a differential that denotes the reaction occurring once (dξ). At constant pressure and temperature the above equations can be written as
 which is the “Gibbs free energy change for the reaction .
which is the “Gibbs free energy change for the reaction .
This results in:
 .
.
By substituting the chemical potentials:
 ,
,
the relationship becomes:

 :
:
which is the standard Gibbs energy change for the reaction that can be calculated using thermodynamical tables. The reaction quotient is defined as:

Therefore,

At equilibrium:

leading to:

and

Obtaining the value of the standard Gibbs energy change, allows the calculation of the equilibrium constant.
Addition of reactants or products
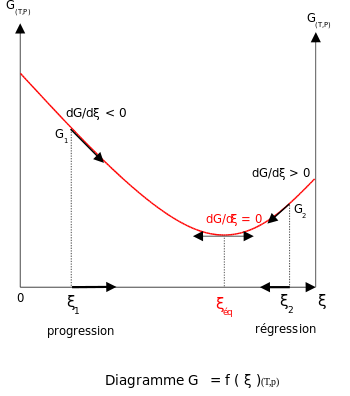
For a reactional system at equilibrium: Qr = Keq; ξ = ξeq.
- If are modified activities of constituents, the value of the reaction quotient changes and becomes different from the equilibrium constant: Qr ≠ Keq

- and

- then

- If activity of a reagent i increases
-
 , the reaction quotient decreases.
, the reaction quotient decreases.
- then
-
 and
and 
- The reaction will shift to the right (i.e. in the forward direction, and thus more products will form).
- If activity of a product j increases
- then
-
 and
and 
- The reaction will shift to the left (i.e. in the reverse direction, and thus less products will form).
Note that activities and equilibrium constants are dimensionless numbers.
Treatment of activity
The expression for the equilibrium constant can be rewritten as the product of a concentration quotient, Kc and an activity coefficient quotient, Γ.
![\displaystyle K={\frac {[\mathrm {S} ]^{\sigma }[\mathrm {T} ]^{\tau }...}{[\mathrm {A} ]^{\alpha }[\mathrm {B} ]^{\beta }...}}\times {\frac {{\gamma _{\mathrm {S} }}^{\sigma }{\gamma _{\mathrm {T} }}^{\tau }...}{{\gamma _{\mathrm {A} }}^{\alpha }{\gamma _{\mathrm {B} }}^{\beta }...}}=K_{\mathrm {c} }\Gamma](https://www.freesandal.org/wp-content/ql-cache/quicklatex.com-9acc2c40aaf5665a61753f0e9d1cd345_l3.png "Rendered by QuickLaTeX.com")
[A] is the concentration of reagent A, etc. It is possible in principle to obtain values of the activity coefficients, γ. For solutions, equations such as the Debye–Hückel equation or extensions such as Davies equation[9] Specific ion interaction theory or Pitzer equations[10]may be used.Software (below) However this is not always possible. It is common practice to assume that Γ is a constant, and to use the concentration quotient in place of the thermodynamic equilibrium constant. It is also general practice to use the term equilibrium constant instead of the more accurate concentration quotient. This practice will be followed here.
For reactions in the gas phase partial pressure is used in place of concentration and fugacity coefficient in place of activity coefficient. In the real world, for example, when making ammonia in industry, fugacity coefficients must be taken into account. Fugacity, f, is the product of partial pressure and fugacity coefficient. The chemical potential of a species in the gas phase is given by

so the general expression defining an equilibrium constant is valid for both solution and gas phases.
Metastable mixtures
A mixture may appear to have no tendency to change, though it is not at equilibrium. For example, a mixture of SO2 and O2 is metastable as there is a kinetic barrier to formation of the product, SO3.
- 2 SO2 + O2 ⇌ 2 SO3
The barrier can be overcome when a catalyst is also present in the mixture as in the contact process, but the catalyst does not affect the equilibrium concentrations.
Likewise, the formation of bicarbonate from carbon dioxide and water is very slow under normal conditions
- CO2 + 2 H2O ⇌ HCO−
3 + H3O+
but almost instantaneous in the presence of the catalytic enzyme carbonic anhydrase.
Pure substances
When pure substances (liquids or solids) are involved in equilibria their activities do not appear in the equilibrium constant[12] because their numerical values are considered one.
Applying the general formula for an equilibrium constant to the specific case of a dilute solution of acetic acid in water one obtains
- CH3CO2H + H2O ⇌ CH3CO2− + H3O+
![\displaystyle K_{\mathrm {c} }={\frac {\mathrm {[{CH_{3}CO_{2}}^{-}][{H_{3}O}^{+}]} }{\mathrm {[{CH_{3}CO_{2}H}][{H_{2}O}]} }}](https://www.freesandal.org/wp-content/ql-cache/quicklatex.com-f8782aa9c8fcfaddc4b3d8e66fadb62d_l3.png "Rendered by QuickLaTeX.com")
For all but very concentrated solutions, the water can be considered a “pure” liquid, and therefore it has an activity of one. The equilibrium constant expression is therefore usually written as
![\displaystyle K={\frac {\mathrm {[{CH_{3}CO_{2}}^{-}][{H_{3}O}^{+}]} }{\mathrm {[{CH_{3}CO_{2}H}]} }}=K_{\mathrm {c} }](https://www.freesandal.org/wp-content/ql-cache/quicklatex.com-40279d14cc0a2aa3cfa87ae20d373fe9_l3.png "Rendered by QuickLaTeX.com") .
.
A particular case is the self-ionization of water itself
- 2 H2O ⇌ H3O+ + OH−
Because water is the solvent, and has an activity of one, the self-ionization constant of water is defined as
![\displaystyle K_{\mathrm {w} }=\mathrm {[H^{+}][OH^{-}]}](https://www.freesandal.org/wp-content/ql-cache/quicklatex.com-5f87576e6f6a9194ea72e47047935381_l3.png "Rendered by QuickLaTeX.com")
It is perfectly legitimate to write [H+] for the hydronium ion concentration, since the state of solvation of the proton is constant (in dilute solutions) and so does not affect the equilibrium concentrations. Kw varies with variation in ionic strength and/or temperature.
The concentrations of H+ and OH− are not independent quantities. Most commonly [OH−] is replaced by Kw[H+]−1 in equilibrium constant expressions which would otherwise include hydroxide ion.
Solids also do not appear in the equilibrium constant expression, if they are considered to be pure and thus their activities taken to be one. An example is the Boudouard reaction:[12]
- 2 CO ⇌ CO2 + C
for which the equation (without solid carbon) is written as:
![\displaystyle K_{\mathrm {c} }={\frac {\mathrm {[CO_{2}]} }{\mathrm {[CO]^{2}} }}](https://www.freesandal.org/wp-content/ql-cache/quicklatex.com-5cc68b6ffa6a27239399ec21af4384e4_l3.png "Rendered by QuickLaTeX.com")
Multiple equilibria
Consider the case of a dibasic acid H2A. When dissolved in water, the mixture will contain H2A, HA− and A2−. This equilibrium can be split into two steps in each of which one proton is liberated.
![\displaystyle {\begin{array}{rl}{\ce {H2A <=> HA^- + H+}}:&K_{1}={\frac {{\ce {[HA-][H+]}}}{{\ce {[H2A]}}}}\\{\ce {HA- <=> A^2- + H+}}:&K_{2}={\frac {{\ce {[A^{2-}][H+]}}}{{\ce {[HA-]}}}}\end{array}}](https://www.freesandal.org/wp-content/ql-cache/quicklatex.com-378cdc06d6b88b3142e05d1c302fd195_l3.png "Rendered by QuickLaTeX.com")
K1 and K2 are examples of stepwise equilibrium constants. The overall equilibrium constant, βD, is product of the stepwise constants.
 :
: ![\displaystyle \beta _{{\ce {D}}}={\frac {{\ce {[A^{2-}][H^+]^2}}}{{\ce {[H_2A]}}}}=K_{1}K_{2}](https://www.freesandal.org/wp-content/ql-cache/quicklatex.com-79650d23a25424d68e8db2ff08f41d20_l3.png "Rendered by QuickLaTeX.com")
Note that these constants are dissociation constants because the products on the right hand side of the equilibrium expression are dissociation products. In many systems, it is preferable to use association constants.
![\displaystyle {\begin{array}{ll}{\ce {A^2- + H+ <=> HA-}}:&\beta _{1}={\frac {{\ce {[HA^-]}}}{{\ce {[A^{2-}][H+]}}}}\\{\ce {A^2- + 2H+ <=> H2A}}:&\beta _{2}={\frac {{\ce {[H2A]}}}{{\ce {[A^{2-}][H+]^2}}}}\end{array}}](https://www.freesandal.org/wp-content/ql-cache/quicklatex.com-4791ebc13fa560a2a0c3af5576b73f35_l3.png "Rendered by QuickLaTeX.com")
β1 and β2 are examples of association constants. Clearly β1 = 1/K2 and β2 = 1/βD; log β1 = pK2 and log β2 = pK2 + pK1[13] For multiple equilibrium systems, also see: theory of Response reactions.
Effect of temperature
The effect of changing temperature on an equilibrium constant is given by the van ‘t Hoff equation

- Thus, for exothermic reactions (ΔH is negative), K decreases with an increase in temperature, but, for endothermic reactions, (ΔH is positive) K increases with an increase temperature. An alternative formulation is

At first sight this appears to offer a means of obtaining the standard molar enthalpy of the reaction by studying the variation of K with temperature. In practice, however, the method is unreliable because error propagation almost always gives very large errors on the values calculated in this way.