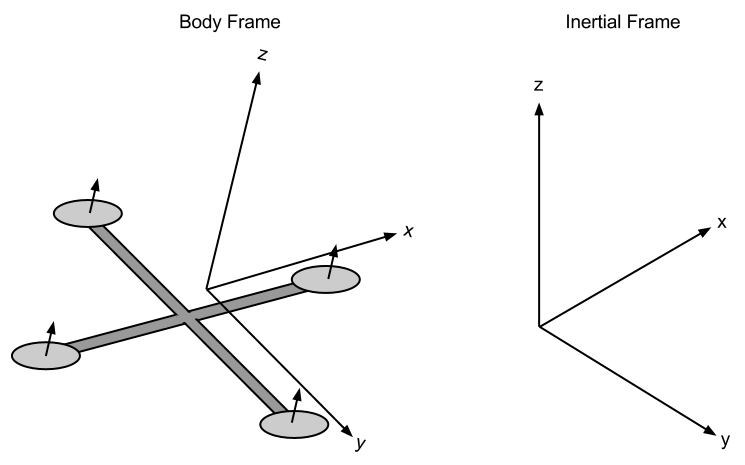
所以能明白 Michael Nielsen 先生為何會突起『費米』之『大象說』的了︰
Overfitting and regularization
The Nobel prizewinning physicist Enrico Fermi was once asked his opinion of a mathematical model some colleagues had proposed as the solution to an important unsolved physics problem. The model gave excellent agreement with experiment, but Fermi was skeptical. He asked how many free parameters could be set in the model. “Four” was the answer. Fermi replied*
*The quote comes from a charming article by Freeman Dyson, who is one of the people who proposed the flawed model. A four-parameter elephant may be found here.
: “I remember my friend Johnny von Neumann used to say, with four parameters I can fit an elephant, and with five I can make him wiggle his trunk.”.
The point, of course, is that models with a large number of free parameters can describe an amazingly wide range of phenomena. Even if such a model agrees well with the available data, that doesn’t make it a good model. It may just mean there’s enough freedom in the model that it can describe almost any data set of the given size, without capturing any genuine insights into the underlying phenomenon. When that happens the model will work well for the existing data, but will fail to generalize to new situations. The true test of a model is its ability to make predictions in situations it hasn’t been exposed to before.
Fermi and von Neumann were suspicious of models with four parameters. Our 30 hidden neuron network for classifying MNIST digits has nearly 24,000 parameters! That’s a lot of parameters. Our 100 hidden neuron network has nearly 80,000 parameters, and state-of-the-art deep neural nets sometimes contain millions or even billions of parameters. Should we trust the results?
───
還特意給了個鍊結
How to fit an elephant
John von Neumann famously said
With four parameters I can fit an elephant, and with five I can make him wiggle his trunk.
By this he meant that one should not be impressed when a complex model fits a data set well. With enough parameters, you can fit any data set.
It turns out you can literally fit an elephant with four parameters if you allow the parameters to be complex numbers.
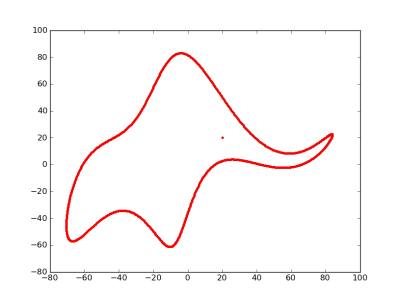
I mentioned von Neumann’s quote on StatFact last week and Piotr Zolnierczuk replied with reference to a paper explaining how to fit an elephant:
“Drawing an elephant with four complex parameters” by Jurgen Mayer, Khaled Khairy, and Jonathon Howard, Am. J. Phys. 78, 648 (2010), DOI:10.1119/1.3254017.
Piotr also sent me the following Python code he’d written to implement the method in the paper. This code produced the image above.
───
大概不會是潛意識裡希望『拯救大象』的吧??又怎知卻挑起人對『大象林旺』之幽思呢!!
─── 《W!O+ 的《小伶鼬工坊演義》︰神經網絡【轉折點】一》
『物質』若是不『穩定』,這個世界將成何面貌?
『物質』若是不『變化』,宇宙哪裡會有生命耶??
『造物』自然也『定理』,玄機一時難釐清!
Chemical bond
A chemical bond is a lasting attraction between atoms, ions or molecules that enables the formation of chemical compounds. The bond may result from the electrostatic force of attraction between oppositely charged ions as in ionic bonds or through the sharing of electrons as in covalent bonds. The strength of chemical bonds varies considerably; there are “strong bonds” or “primary bonds” such as covalent, ionic and metallic bonds, and “weak bonds” or “secondary bonds” such as dipole–dipole interactions, the London dispersion force and hydrogen bonding.
Since opposite charges attract via a simple electromagnetic force, the negatively charged electrons that are orbiting the nucleus and the positively charged protons in the nucleus attract each other. An electron positioned between two nuclei will be attracted to both of them, and the nuclei will be attracted toward electrons in this position. This attraction constitutes the chemical bond. Due to the matter wave nature of electrons and their smaller mass, they must occupy a much larger amount of volume compared with the nuclei, and this volume occupied by the electrons keeps the atomic nuclei in a bond relatively far apart, as compared with the size of the nuclei themselves.
In general, strong chemical bonding is associated with the sharing or transfer of electrons between the participating atoms. The atoms in molecules, crystals, metals and diatomic gases—indeed most of the physical environment around us—are held together by chemical bonds, which dictate the structure and the bulk properties of matter.

Examples of Lewis dot-style representations of chemical bonds between carbon (C), hydrogen (H), and oxygen (O). Lewis dot diagrams were an early attempt to describe chemical bonding and are still widely used today.
All bonds can be explained by quantum theory, but, in practice, simplification rules allow chemists to predict the strength, directionality, and polarity of bonds. The octet rule and VSEPR theory are two examples. More sophisticated theories are valence bond theory which includes orbital hybridization and resonance, and molecular orbital theory which includes linear combination of atomic orbitals and ligand field theory.Electrostatics are used to describe bond polarities and the effects they have on chemical substances.
『大象』四輔可定形,十三『參數』恐太多!!
History
Early speculations about the nature of the chemical bond, from as early as the 12th century, supposed that certain types of chemical species were joined by a type of chemical affinity. In 1704, Sir Isaac Newton famously outlined his atomic bonding theory, in “Query 31” of his Opticks, whereby atoms attach to each other by some “force“. Specifically, after acknowledging the various popular theories in vogue at the time, of how atoms were reasoned to attach to each other, i.e. “hooked atoms”, “glued together by rest”, or “stuck together by conspiring motions”, Newton states that he would rather infer from their cohesion, that “particles attract one another by some force, which in immediate contact is exceedingly strong, at small distances performs the chemical operations, and reaches not far from the particles with any sensible effect.”
In 1819, on the heels of the invention of the voltaic pile, Jöns Jakob Berzelius developed a theory of chemical combination stressing the electronegative and electropositive characters of the combining atoms. By the mid 19th century, Edward Frankland, F.A. Kekulé, A.S. Couper, Alexander Butlerov, and Hermann Kolbe, building on the theory of radicals, developed the theory of valency, originally called “combining power”, in which compounds were joined owing to an attraction of positive and negative poles. In 1916, chemistGilbert N. Lewis developed the concept of the electron-pair bond, in which two atoms may share one to six electrons, thus forming the single electron bond, a single bond, a double bond, or a triple bond; in Lewis’s own words, “An electron may form a part of the shell of two different atoms and cannot be said to belong to either one exclusively.”[2]
That same year, Walther Kossel put forward a theory similar to Lewis’ only his model assumed complete transfers of electrons between atoms, and was thus a model of ionic bonding. Both Lewis and Kossel structured their bonding models on that of Abegg’s rule(1904).
Niels Bohr proposed a model of the atom and a model of the chemical bond. According to his model for a diatomic molecule, the electrons of the atoms of the molecule form a rotating ring whose plane is perpendicular to the axis of the molecule and equidistant from the atomic nuclei. The dynamic equilibrium of the molecular system is achieved through the balance of forces between the forces of attraction of nuclei to the plane of the ring of electrons and the forces of mutual repulsion of the nuclei. The Bohr model of the chemical bond took into account the Coulomb repulsion – the electrons in the ring are at the maximum distance from each other.[3][4]
In 1927, the first mathematically complete quantum description of a simple chemical bond, i.e. that produced by one electron in the hydrogen molecular ion, H2+, was derived by the Danish physicist Oyvind Burrau.[5] This work showed that the quantum approach to chemical bonds could be fundamentally and quantitatively correct, but the mathematical methods used could not be extended to molecules containing more than one electron. A more practical, albeit less quantitative, approach was put forward in the same year byWalter Heitler and Fritz London. The Heitler–London method forms the basis of what is now called valence bond theory. In 1929, the linear combination of atomic orbitals molecular orbital method (LCAO) approximation was introduced by Sir John Lennard-Jones, who also suggested methods to derive electronic structures of molecules of F2 (fluorine) and O2 (oxygen) molecules, from basic quantum principles. This molecular orbital theory represented a covalent bond as an orbital formed by combining the quantum mechanicalSchrödinger atomic orbitals which had been hypothesized for electrons in single atoms. The equations for bonding electrons in multi-electron atoms could not be solved to mathematical perfection (i.e., analytically), but approximations for them still gave many good qualitative predictions and results. Most quantitative calculations in modern quantum chemistry use either valence bond or molecular orbital theory as a starting point, although a third approach, density functional theory, has become increasingly popular in recent years.
In 1933, H. H. James and A. S. Coolidge carried out a calculation on the dihydrogen molecule that, unlike all previous calculation which used functions only of the distance of the electron from the atomic nucleus, used functions which also explicitly added the distance between the two electrons.[6] With up to 13 adjustable parameters they obtained a result very close to the experimental result for the dissociation energy. Later extensions have used up to 54 parameters and gave excellent agreement with experiments. This calculation convinced the scientific community that quantum theory could give agreement with experiment. However this approach has none of the physical pictures of the valence bond and molecular orbital theories and is difficult to extend to larger molecules.
五四實繪不存疑??!!
Theories of chemical bonding

In the (unrealistic) limit of “pure” ionic bonding, electrons are perfectly localized on one of the two atoms in the bond. Such bonds can be understood by classical physics. The forces between the atoms are characterized by isotropic continuum electrostatic potentials. Their magnitude is in simple proportion to the charge difference.
Covalent bonds are better understood by valence bond theory or molecular orbital theory. The properties of the atoms involved can be understood using concepts such as oxidation number. The electron density within a bond is not assigned to individual atoms, but is instead delocalized between atoms. In valence bond theory, the two electrons on the two atoms are coupled together with the bond strength depending on the overlap between them. In molecular orbital theory, the linear combination of atomic orbitals (LCAO) helps describe the delocalized molecular orbital structures and energies based on the atomic orbitals of the atoms they came from. Unlike pure ionic bonds, covalent bonds may have directed anisotropic properties. These may have their own names, such as sigma bond and pi bond.
In the general case, atoms form bonds that are intermediate between ionic and covalent, depending on the relative electronegativity of the atoms involved. This type of bond is sometimes called polar covalent.
傳說
一個和尚提水喝;兩個和尚抬水喝;三個和尚沒水喝。
似乎『巨觀說法』早登台!!??
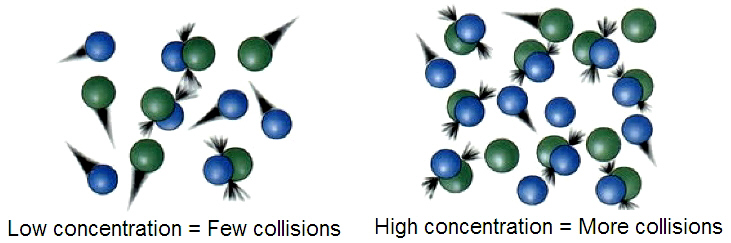
化學勢
在熱力學中,某種物質的化學勢指的是,在化學反應或者相變中,此物質的粒子數發生改變時所吸收或放出的能量。在混合物中的某種物質的化學勢定義為此熱力學系統的吉布斯自由能對此物質粒子數的變化率,即偏導數(其他物質的粒子數及其他系統參數保持不變)。當溫度和壓力固定時,化學勢也被稱作偏莫耳吉布斯自由能 ,或者莫耳化學勢[1]。在化學平衡或相平衡狀態下,自由能處於極小值,各種物質的化學勢與化學計量係數乘積之加和為零[2][3][4]。
概述
粒子總是趨向於從高化學勢流向低化學勢,因而,化學勢可視為物理中「位能」概念的推廣。當一個球從山上滾下,它從高重力勢(有更多的做功「趨勢」)跑到了低重力勢的區域。同樣,分子在運動、發生化學反應、溶解、融化等過程中,它們總是趨向於自發的從高化學勢的狀態變到低化學勢的狀態,相應的,此分子的的粒子數會發生變化,而粒子數是化學勢的共軛變量。
一個簡單的例子是,一個稀疏氣體分子體系在一個均勻環境中的擴散過程。在這個過程中,分子會自發的從高密度分布區域擴散到低密度分布的區域,直到此分子在各處的分布密度都相等。
我們可以用機械運動的理論對分子的隨機運動做微觀解釋,但是,如果用化學勢來理解這個過程則顯得更加簡單方便:在確定的溫度下,一個分子在高密度區域有更高的化學勢,而在低密度區域的化學勢則較低。當分子從高化學勢區域流到低化學勢區域時,就會釋放自由能,因此這是一個自發的過程。
另一個例子是相變過程。我們可以認為,在0°C以上時,水分子的液相(液態水)具有更低的化學勢,而固相(即冰)化學勢更高,冰化為水則是從高化學勢變為低化學勢,而在0°C以下時則正好相反。而正好處於0 °C的冰水混合物,固相和液相的化學勢則是正好相等的,系統處於平衡。
另外再比如水溶液中弱酸的解離過程,比如乙酸,HA(A=CH3COO−):
- HA ⇌ H+ + A−
醋中就包含乙酸。當乙酸分子解離時,未解離的乙酸分子數量減少 ,而產物離子(H+和A−)數量增加。在此過程中,乙酸分子的化學勢變小,而H+和A−離子的化學勢升高,當反應物與產物的化學勢相等時,系統達到平衡。
化學勢的概念被運用於很多關於化學平衡的方面,比如熔化、沸騰 、蒸發、溶解、滲透、分配係數、液體萃取、層析分離,這些情況下通常都會有一個表徵各分量物質化學勢的函數。
在電化學中,離子不一定總是從高化學勢流向低化學勢,但是它們會從高的「電化學勢」流向低的「電化學勢」,與化學勢不同的是 ,電化學勢還計入了靜電力的作用,因而可以完全的描述離子的運動。
熱力學的定義
某種粒子-i(原子、分子或原子核)的化學勢  是一個強度量,其定義來自於唯象引入的的熱力學基本關係[6]
是一個強度量,其定義來自於唯象引入的的熱力學基本關係[6]

這裡  是系統內能 U 的變化微元,
是系統內能 U 的變化微元,  是系統熵 S 的變化,
是系統熵 S 的變化,  是體積的變化,而
是體積的變化,而  是第i種粒子的粒子數
是第i種粒子的粒子數  的變化, T 是絕對溫度,P是壓力。當存在電磁場時,此式中還要計入相應的電磁做功的項。
的變化, T 是絕對溫度,P是壓力。當存在電磁場時,此式中還要計入相應的電磁做功的項。
由上述熱力學基本關係,我們得到化學勢的定義為:

然而此定義對於實際系統有一些不方便,比如說對於化學溶液,當加入粒子時,又要想保持體積和熵等變量不變,這幾乎是不可能的。為此,我們通過Legendre變換來引入另一個熱力勢:吉布斯自由能,  ,將此形式微分得到:
,將此形式微分得到:  ,將這裡的dU用上面的熱力學基本關係替換,我們就可以得到:
,將這裡的dU用上面的熱力學基本關係替換,我們就可以得到:

於是,我們就得到化學勢 的另一個表達式:

在改變吉布斯自由能時同時又固定溫度和壓力不變則是在通常情況下可以做到的,於是,這種情況下我們可以得到

若一個系統的溫度和壓力固定不變,而又可以與外界環境有粒子交換,熱力學平衡狀態就意味著系統的吉布斯自由能應當處於極小值,即:

這一等式關係可以用於建立化學反應的平衡常數。
類似的,我們還可以對 做其它形式的 Legendre變換,從而得到其它的熱力學勢函數,如焓  , Helmholtz自由能
, Helmholtz自由能  ,於是相應的化學勢為:
,於是相應的化學勢為:

這些不同的形式都是化學勢,只是運用於不同的物理條件下。
應用
Gibbs–Duhem方程式描述了一個熱力學系統中的組分的化學勢變化之間的關係。例如一種由兩種物質組成的混合物,在確定的溫度和壓力下,這兩種物質的化學勢滿足如下關係:

相變平衡或化學平衡往往都會有一個決定相變的常數。比如,冰融化的溫度(冰點),即為固相和液相平衡的溫度。化學勢可以用來解釋由Clapeyron方程式解出的相圖中的斜率[7],這些也可以由Gibbs–Duhem方程式給出,還可以解釋一些「依數性」(如溶劑蒸氣壓的降低導致溶液凝固點下降)[8],並可以很自然的導出拉午耳定律和亨利定律[9]。
歷史
化學勢首先由美國工程師、化學家、數學物理學家吉布斯(Josiah Willard Gibbs)提出:
向處於靜壓力狀態的某種均勻物質中加入無窮小量的任意添加物,而保持其仍然分布均勻且熵和體積都不變,則此時總能量變化量對於添加物粒子數的微商表徵了一種添加這種組分的位能。
“If to any homogeneous mass in a state of hydrostatic stress we suppose an infinitesimal quantity of any substance to be added, the mass remaining homogeneous and its entropy and volume remaining unchanged, the increase of the energy of the mass divided by the quantity of the substance added is the potential for that substance in the mass considered.”
吉布斯隨後意識到,由此定義出發,新加入體系的物質不一定需要是在原體系中已經存在的物質,而可以是任何化學成分,也可以是比例確定的混合成分,這種自由度可以使化學勢應用於熱力學和物理學中各種正經歷物質成分變化的系統。化學勢也被稱為偏莫耳吉布斯自由能(或偏莫耳性質),單位為能量/粒子數,或能量/莫耳 。
1873年,吉布斯在他的論文《物質熱力學性質的幾何面表示法》(A Method of Geometrical Representation of the Thermodynamic Properties of Substances by Means of Surfaces)中,提出了他的新方程式及其基本原理,用於討論當不同系統相互接觸時發生的自發過程。對於相互接觸的均勻物質(如固、液、氣組分),通過三維的體積-熵-內能圖,吉布斯定義了三種狀態:「必要穩定(necessarily stable)」,「中性(neutral)」,「不穩定(unstable)」,並借之理解某個變化過程能否自發進行。1876年,吉布斯在此理論基礎之上引入了化學勢的概念用於理解化學反應過程。他對此總結道:
考慮一種物質,處於具有確定壓力和溫度的某介質中,那麼此物質處於熱力學平衡的充要條件是:
其中
表示由系統各組分狀態變化或比例變化造成的變分量。穩定平衡的條件是,上面括號中的表達式取極小值。
“If we wish to express in a single equation the necessary and sufficient condition of thermodynamic equilibrium for a substance when surrounded by a medium of constant pressure P and temperature T, this equation may be written:
where δ refers to the variation produced by any variations in the state of the parts of the body, and (when different parts of the body are in different states) in the proportion in which the body is divided between the different states. The condition of stable equilibrium is that the value of the expression in the parenthesis shall be a minimum.”

注意這裡與現代常用的符號不同的是,吉布斯用  表示內能,
表示內能,  表示熵,
表示熵,  表示系統體積。
表示系統體積。


 and
and  in a schematic example below) by the appropriate integers a, b, c and d.
in a schematic example below) by the appropriate integers a, b, c and d.




 .
.



![\displaystyle v=-{\frac {d[{\ce {A}}]}{dt}}=k\cdot [{\ce {A}}].](http://www.freesandal.org/wp-content/ql-cache/quicklatex.com-48ec258f44075e367017ff93506461d2_l3.png "Rendered by QuickLaTeX.com")
![\displaystyle {\ce {[A]}}(t)={\ce {[A]}}_{0}\cdot e^{-k\cdot t}.](http://www.freesandal.org/wp-content/ql-cache/quicklatex.com-346e4a7e4821550de620ad417c196e0f_l3.png "Rendered by QuickLaTeX.com")








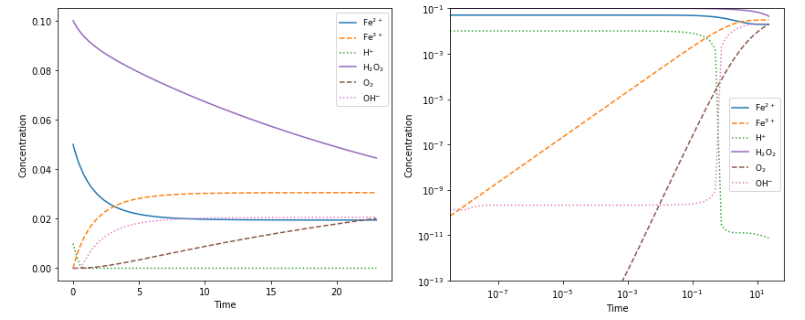
 molecules. The sheer number allows us to describe this inherently stochastic process deterministically.
molecules. The sheer number allows us to describe this inherently stochastic process deterministically.
 is given by:
is given by:
 is a matrix with the overall net stoichiometric coefficients (positive for net production, negative for net consumption), and
is a matrix with the overall net stoichiometric coefficients (positive for net production, negative for net consumption), and  is a matrix with the multiplicities of each reactant for each equation.
is a matrix with the multiplicities of each reactant for each equation.

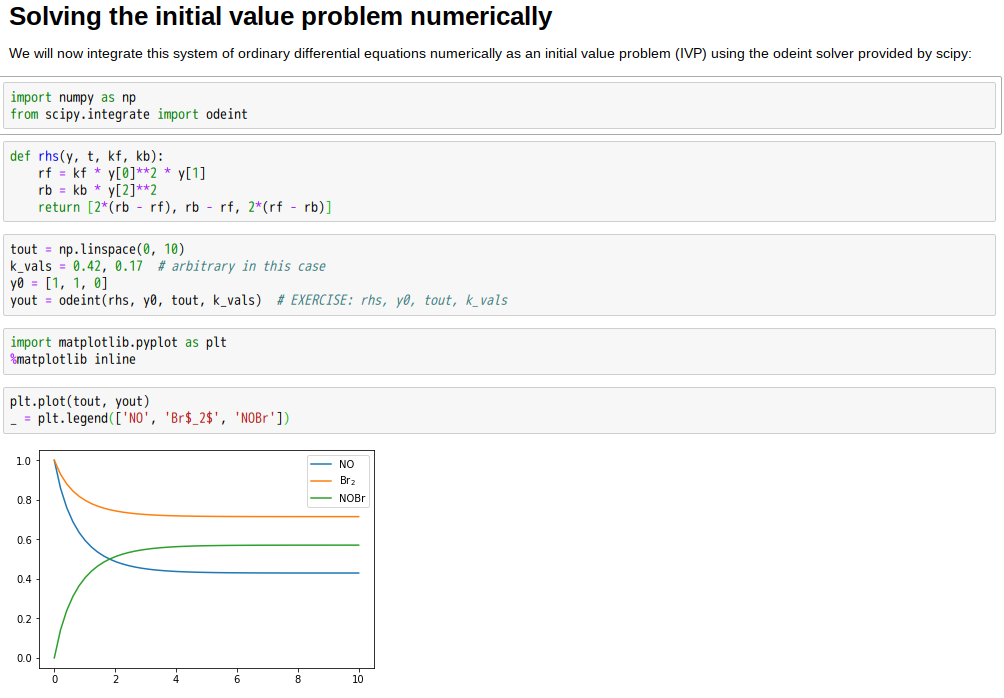
 and the square of the concentration of NO. The rate of the second reaction (the backward process) is in analogy proportional to the square of the concentration of NOBr. Using the proportionality constants
and the square of the concentration of NO. The rate of the second reaction (the backward process) is in analogy proportional to the square of the concentration of NOBr. Using the proportionality constants  and
and  we can formulate our system of nonlinear ordinary differential equations as follows:
we can formulate our system of nonlinear ordinary differential equations as follows:
 ,
,  ,
,  respectively.
respectively.








 ,
, ,
,
![\left[I_m(\cdots, t) \Rightarrow_{S} O_m(\cdots, t)\right] \Rightarrow_{S} \left[I_m(\cdots, t + \tau) \Rightarrow_{S} O_m(\cdots, t + \tau)\right]](http://www.freesandal.org/wp-content/ql-cache/quicklatex.com-4fdacc020a4c6714fc36227ae1061aa3_l3.png "Rendered by QuickLaTeX.com")











 和初始時間
和初始時間  的函數,可以將時間
的函數,可以將時間  和此矩陣相乘,得到時間
和此矩陣相乘,得到時間 
 為系統狀態,
為系統狀態,  為輸入信號,而
為輸入信號,而  為時間
為時間  ,其解如下
,其解如下
 定義為
定義為